Posted on Tuesday, March 05, 2013 at 11:28AM by The GenePattern Team
Overview
After aligning and/or assembling your RNA-seq data, it is important to take a closer look at the content of those result files before continuing with further analysis; in part, because the results of that investigation may, in fact, point you toward how you should best analyze your data.
Specifically in GenePattern, modules are provided to calculate such Quality Control (QC) metrics as: Depth of Coverage, Continuity of Coverage, Duplication Rate, Expression Rates, Strand Specificity, and GC content, among others.
Having these sorts of metrics can help to prevent or better understand common RNA-seq errors stemming from such sources as: read length, quality of data, sample prep, or number of reads in the data.
Modules in GP
- Picard.AddOrReplaceReadGroups
- SortSam
- Picard.CreateSequenceDictionary
- Picard.ReorderSam
- Picard.MarkDuplicates
- SAMtools.FastaIndex
- RNAseqMetrics
The following decision diagram illustrates a suggested workflow. This workflow is discussed in further detail in subsequent sections.
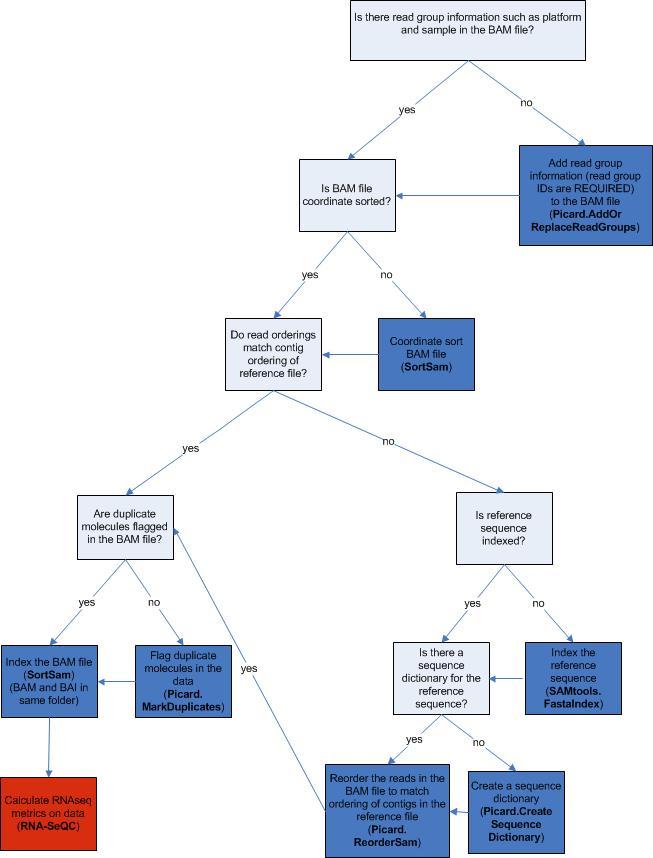
Input
The input to the suggested RNA-seq QC workflow in GenePattern is an aligned, coordinate sorted BAM file with Read Group information (such as platform or sample) in the header. (SAM files can be converted to BAM format using the SortSam module.)
If your aligned BAM file does not contain Read Group Information you should run your data through AddOrReplaceReadGroups, as discussed next.
If the aligned BAM file is not coordinate sorted, run the data through SortSam, making sure the sort order is set to "coordinate" as discussed below.
More information about the SAM/BAM format can be found at the SAMtools website.
Note that if your data was aligned with TopHat, you will likely want to run your BAM file through the Picard tool MergeBamAlignment (soon to be available in GenePattern) to handle the fact that TopHat removes unaligned reads. This can throw off the total number of reads and any other metrics using that value in the RNAseqMetrics module.
1) Picard.AddOrReplaceReadGroups
Input for Picard.AddOrReplaceReadGroups is a BAM file which has been aligned.
The module will either add new (if none previously existed) or replace read groups as defined in the parameters. All reads in the file will be assigned to the specified read group.
Read Group information is required by Picard.MarkDuplicates and the RNAseqMetrics module. Specifically the RNAseqMetrics module requires a Read Group ID in the BAM header.
Full documentation for Picard.AddOrReplaceReadGroups, with parameter descriptions, is available here.
2) SortSam
SortSam takes as input a BAM file and outputs a sorted and indexed file. In this step of the workflow, the input BAM should come from Picard.AddOrReplaceReadGroups in step 1.
Note that the module will only generate an index if the output file time is "BAM" and the sort order is "coordinate".
Full documentation for SortSam can be found here.
3) SAMtools.FastaIndex
SAMtools.FastaIndex takes a reference FASTA (.fa) file and creates a .fai index file for it, which will be used by both Picard.ReorderSam, in step 5, and RNAseqMetrics, in step 8, to quickly locate and retrieve information from the reference sequence.
Full documentation for SAMtools.FastaIndex can be found here.
4) Picard.CreateSequenceDictionary
Next Picard.CreateSequenceDictionary takes a reference FASTA (.fa) and creates a SAM file containing a sequence dictionary (.dict extension). Sequence dictionaries contain the sequence name, length and genome assembly identifier and other information about sequences. The .dict file is required for both Picard.ReorderSam, in step 5, and RNAseqMetrics, in step 8.
The output FASTA file (.fa) from SAMtools.FastaIndex (step 3), can be passed as input to this module.
Full documentation for Picard.CreateSequenceDictionary can be found here.
5) Picard.ReorderSam
Now that the the .fai and .dict files have been created for the reference FASTA file (steps 3 and 4), Picard.ReorderSam can be run to order the reads in the BAM file according the contigs of a reference FASTA file.
Picard.ReorderSam takes bam/bai pair (for instance, as output by SortSam earlier in this workflow) a.dict from Picard.CreateSequenceDictionary (step 4), and the .fa and .fai files from SAMtools.FastaIndex (step 3) and reorders the BAM file in accordance with the contigs in the reference FASTA file provided. The order is determined by exact name matching of contigs. Reads mapped to contigs absent in the reference file are dropped.
The resulting BAM file can next be sent to Picard.MarkDuplicates.
Full documentation for Picard.ReorderSam can be found here.
6) Picard.MarkDuplicates
Next, Picard.MarkDuplicates takes the coordinate sorted BAM file output by Picard.ReorderSam, in step , (with read group information, added by Picard.AddOrReplaceGroups in step 1).
This is an optional module that will mark duplicate reads in the BAM file and optionally remove them. To see metrics for Duplication rates in the results from RNAseqMetrics, run this module and do not remove the duplicate reads.
Full documentation for Picard.MarkDuplicates can be found here.
7) SortSam
The last step before running RNAseqMetrics is to index the BAM file which resulted from the workflow above. To do this, run SortSam, selecting BAM as the output format.
8) RNAseqMetrics
RNAseqMetrics is the last step in the GenePattern RNA-seq QC workflow. The module calculates standard RNA-seq related metrics, such as depth of coverage, ribosomal RNA contamination, continuity of coverage, and GC bias. It takes the following as input:
- A coordinate sorted BAM file, and its associated index*, containing a Read Group ID, which has been reordered by contig and optionally has duplicate genes marked. (output by SortSam in step 7)
- A reference FASTA (.fa)*
- The reference FASTA file index (.fai)* (output by SAMtools.FastaIndex in step 3)
- The reference FASTA file dictionary (.dict)* (output by Picard.CreateSequenceDictionary in step 4)
*Note that in most cases these will all need to be specified separately.
- A text formatted sample information file is optional and can be used to label the samples in the output files.
- A gtf annotation file is also required. hg18 and hg19 are provided in a drop down list, other genomes must be provided by the user.
Please read the RNAseqMetrics module documentation for complete information regarding optional input files, parameter settings and the various metrics which will or won't be output based on those settings.
*If using the output from SortSam the BAM and BAI are located in the same folder and only the BAM file need be passed as an input parameter.
Output
The output of the RNAseqMetrics module (and thus this workflow) is a ZIP archive containing an HTML report of metrics stating the total number of reads, depth of coverage at the 3’ and 5’ end, etc. The report also links to a GCT file containing the calculated RPKM values for each transcript in each sample.
Other metrics calculated include:
- total read number
- number of unique reads
- number of duplicate reads (this metric is only calculated if duplicates were marked in the input BAM file(s))
- duplication rate (number of duplicates/total reads) (this metric is only calculated if duplicates were marked in the input BAM file(s))
- number of reads mapped/aligned
- mapping rate (mapped reads/total reads)
- number of unique reads mapped
- mapped unique rate (mapped unique reads/mapped reads)
- reads that are mapped to rRNA regions
- rRNA rate (reads mapped to rRNA regions/mapped reads)
- unique reads mapped to non-rRNA regions
- unique non-rRNA rate (unique reads mapped to non-rRNA regions/mapped reads)
- intragenic rate (reads mapped to intragenic regions/mapped unique reads)
- exonic rate (reads mapped to exonic regions/mapped unique reads)
- coding rate (reads mapped to coding regions/mapped unique reads)
- intron or UTR rate (reads mapped to intronic or UTR regions/mapped unique reads)
- intergenic rate (reads mapped to intergenic regions/mapped unique reads)
- strand specificity (this metric is only calculated for paired-end BAM files)
- coverage metrics (particularly for the top four expressed transcripts)
- RPKM: this metric quantifies transcript levels in reads per kilobase of exon model per million mapped reads (RPKM). The RPKM measure of read density reflects the molar concentration of a transcript in the starting sample by normalizing for RNA length and for the total read number in the measurement. This facilitates transparent comparison of transcript levels both within and between samples.
- exon reads, exon length, intron reads, intron length, and related numbers for each transcript
Full documentation for the module and its output files can be found here: (give public link when ready)
Note
This workflow, and specifically the RNAseqMetrics module, has been optimized for Eukaryotic RNA-seq data. Modules which comprise methods optimized for Prokaryotic data are currently not available.